Prolog
The following text gives some basic insight into XAS/XES. Scope and references are incomplete but I hope some readers will find ideas for research. I thank Janine Grattage, Kristina Kvashnina, Uwe Bergmann, Frank de Groot and Steve Cramer who in one way or another contributed. Please contact me in case you find mistakes, would like to propose improvements, have questions or flat out disagree with what I wrote. I hope this page will grow in the future so please come and visit from time to time.
General
Energy analysis of the scattered photons ω in addition to control of the incident photon energy Ω (cf. Figure 1) extends the range of applications of XAS and addresses some problems that XAS cannot solve [1,2]. The instrumental energy bandwidth in XAS is of the order of the core hole lifetime broadening, which is around 1 eV for 3d transition metal (TM) K edges and as large as 8 eV at the L-edge of actinides. Hard X-ray monochromators are typically based on perfect-crystal Bragg optics for the incident photons and the same principle can be used to analyze the scattered photons. Various geometries using spherical, cylindrical and flat crystals with point-to-point or dispersive geometries have been used at synchrotron radiation beamlines (for references see Synchrotron Radiation News (2009) 22 12-16). Figure 2 shows a point-to-point Rowland geometry with five spherically bent crystals.

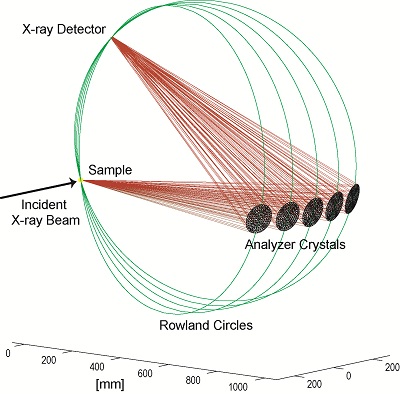
Even though the energy bandwidth is defined by the analyzers (intrinsic and geometry contributions), often energy dispersive detectors are used for photon counting in order to suppress background from unwanted X-ray events (scattered X-rays that somehow find their way into the detector or Bragg reflections from the analyzers of different orders).(Cat. Tod. 145 294-299 (2009))
A number of spectroscopic techniques become available using such an experimental setup. They can be grouped into the following categories:
I. High-Energy Resolution Fluorescence Detected (HERFD) XAS
II. Non-Resonant X-Ray Emission Spectroscopy (XES)
III. Resonant X-Ray Emission Spectroscopy (RXES) and Resonant Inelastic X-ray Scattering
(RIXS)
IV. X-Ray Raman Spectroscopy (XRS) (check excellent
intro on wikipedia)
The first two techniques complement and extend the current standard XAS experiments while the last two open up possibilities for detailed studies of the electronic structure using a photon-in photon-out technique. The hard X-ray probe makes all techniques suitable for in-situ ( operando ) studies and experiments under extreme conditions (e.g. high pressure).
I. High-Energy Resolution Fluorescence Detected (HERFD) XAS
Experiment: The emitted energy ω is tuned to a fluorescence line and the incident energy Ω is scanned through an absorption edge. The intensity variation of the fluorescence line is recorded as a function of the incident energy.
Fluorescence detected XAS takes advantage of a secondary process - the radiative decay of photoexcited states - which is, in the hard X-ray range, to a very good approximation proportional to the absorption coefficient. (Note that this is not necessarily the case and any secondary process detection is not a true measurement of the absorption coefficient.) An energy dispersive solid state detector provides an energy bandwidth of about 200-300 eV at the Fe Kα line. The captured solid angle depends on the number of elements used and the distance from the sample and can be as high as 0.4 sr. Non-linearity at high count-rates often limits the use of solid state detectors at high flux synchrotron radiation beamlines.
An X-ray spectrometer allows the X-ray detector to be chosen freely because the energy is selected by means of Bragg optics. This ensures a dramatic reduction of unwanted background radiation (only photons of interest reach the detector) and increases the usable dynamic range. Thus, measurements of up to 107 counts/sec with practically no background counts in the desired fluorescence line become feasible using, e.g., an avalanche photodiode (APD) as the X-ray detector.
The XES solid angle is limited by the geometrical constraints of the emission spectrometer. Current multi-crystal spectrometers reach 0.04-0.10 sr. Also, the reflectivity of the analyzer crystals is currently only at 10%. Developments towards smaller bending radii of the analyzer crystals, and thus solid angles up to 0.3 sr, as well as higher efficiency, will make XES a viable competitor to solid state detectors for many experiments at orders of magnitude better energy resolution. The additional freedom of using a very fast detector is particularly interesting for time-resolved studies.
HERFD XAS offers the best possible signal-to-background ratio because the energy resolution is of the order of the core hole lifetime broadening. An important consequence is the suppression of diffraction peaks in XAS measurements of polycrystalline samples. The ideal separation of fluorescence lines in XES furthermore allows the recording of EXAFS beyond unwanted absorption edges in dilute samples [3], e.g. Mn EXAFS in the presence of Fe [4]. The spectral features in HERFD XAS can be sharper than the core hole lifetime broadening [5]. This can be understood within the framework of resonant inelastic X-ray scattering (RIXS). The K absorption pre-edges in 3d transition metals can thus be better separated from the main edge.
The differences between standard and HERFD XAS have been discussed by Carra et al.[6], who
indicated problems in using this technique when attempting to record a true absorption spectrum, and their ideas
have been illustrated by several other authors [7-9]. It was found that HERFD XAS on 5d transition elements can be
interpreted to good accuracy as XAS spectra measured below the core hole lifetime broadening and the technique has
been employed to study CO adsorption on Pt and to identify partially oxidized Au/Al2O3 as a reaction intermediate
[10-11].
Why HERFD and not PFY (partial fluorescence yield)? The line sharpening effect in HERFD is not a
result of the partial fluorescence detection but due to the fact that the experimental energy bandwidth is similar
to the core hole lifetime broadening. The reader is refereed to references [1] and [8] in order to understand the
role of the lifetime broadening in HERFD and RIXS. A PFY spectrum does not necessarily result in sharper spectral
features. Consider e.g. a fluorescence detected XAFS spectrum measured using a Ge detector. This is
already PFY because the detector selects only part of the fluorescence lines but the spectrum is expected to look
like a transmission XAFS. Note that PFY and HERFD are equivalent to Auger-detected XAFS. Photon-in/photon-out
techniques are obviously blind to the photoelectrons so this channel does not appear in RXES and RIXS planes unlike
in resonant Auger spectroscopy.
II. Non-Resonant XES
Experiment: The incident energy Ω is tuned well above an absorption edge and the emitted energy ω is scanned over the energy range of a fluorescence line.
X-ray emission is a second-order process. First, a core hole is created and the X-rays that are
emitted when this core hole is filled give rise to the X-ray emission or fluorescence (cf. Figure 1). The chemical
sensitivity in XES can be due to two mechanisms [12].
A) If the core hole is replaced by another core hole, e.g. a 3p to 1s (Kβ1,3 and
Kβ’’) transition in a 3d transition metal, the sensitivity to the valence electrons is
indirect. The final state core hole interacts with the valence electrons and this interaction shapes the emission
line. The Kβ main lines, for example, are sensitive to the valence shell spin state via an intra-atomic
exchange interaction. This has been exploited to study water oxidation in photosynthesis [13] and in high pressure
applications in earth science [14]. The Kβ lines can also be used to record spin-selective absorption spectra
providing a unique tool to study the 3d shell configuration [15-17].
B) XES can also be used to probe the valence shell directly. In this case, the energy of the emission
spectrometer is tuned close to the energ difference between the core hole in the intermediate state and the Fermi
level of the system under study. It has been shown that these emission lines are very sensitive to the ligand
orbitals of 3d transition metals and thus the type of ligand can be identified [18]. At the L-edges of 5d
transition metals it is possible to probe the metal d-electrons directly and the technique is therefore less
sensitive to the ligand environment and more sensitive to the metal electron configuration. Valence-to-core XES is
comparable to valence shell photoemission with different selection rules. However, XES is bulk sensitive, strictly
element-selective and has the advantage that no high vacuum is required, while the energy resolution is usually
higher in photoemission experiments.
X-ray emission lines are usually recorded after photoionization where it is well known that strong multi-electron excitations occur. A correct theoretical treatment should therefore include all excited intermediate states. This has been shown by comparing Kβ spectra after photoionization and after a radioactive (K capture) decay [19].
back to topIII. Resonant XES (RXES) and Resonant Inelastic X-Ray Scattering (RIXS)
Experiment: The incident energy Ω is scanned across an absorption edge. The emitted energy ω is also scanned either over the fluorescence lines or over energies just below the elastically scattered peak. In the latter case, the energy transfer Ω-ω becomes small (on the order of a few eV) and valence band excitations are observed.
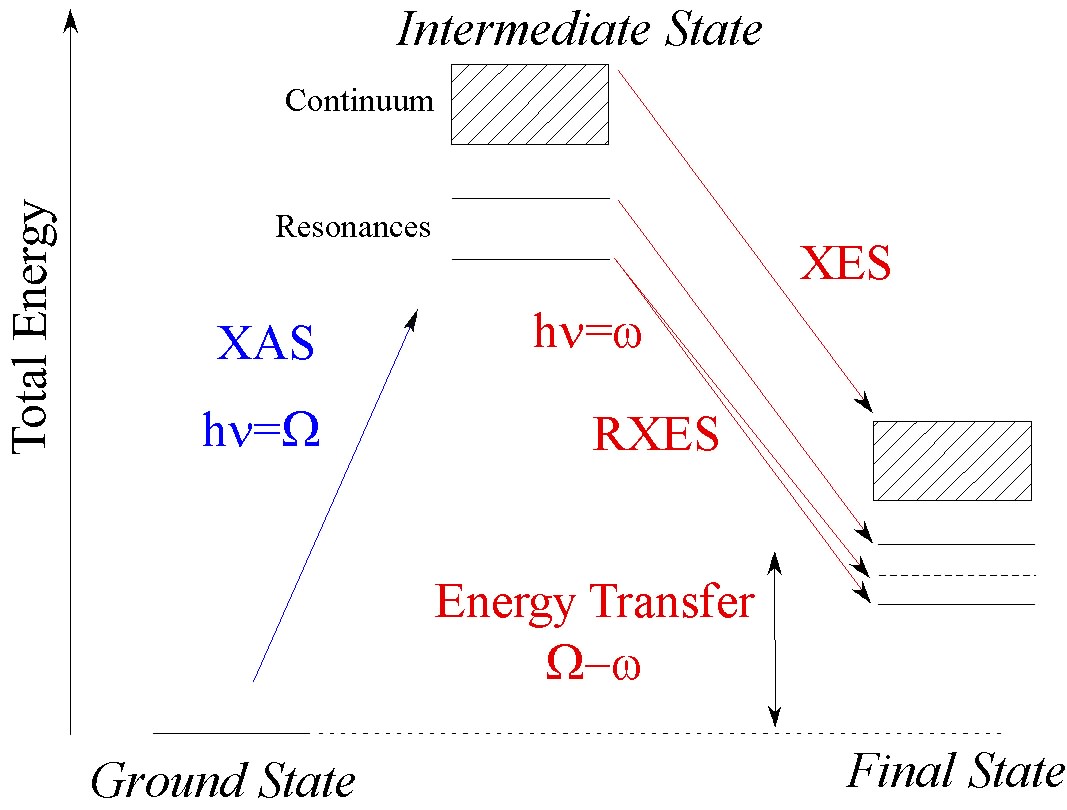
Resonant inelastic X-ray scattering (RIXS) or resonant X-ray emission spectroscopy (RXES) is used to study the electronic structure around a central (X-ray scattering) atom. It requires the incident energy to be close to an absorption edge, i.e. an excitation to a bound state. It is possible to distinguish between two cases of resonant scattering:
- A fluorescence line can be measured after resonant excitation. This is referred to as resonant X-ray emission spectroscopy (RXES) or direct RIXS. In a one-electron picture one could refer to this process as a spectator decay: The photo-excited electron remains in a bound, previously unfilled orbital and an electron from a filled orbital decays to fill the core hole.
- Staying in the one-electron picture: The photo-excited electron decays to fill the core hole (participator decay). The modification of the Coulomb potential due to photo-excitation and decay may give rise to an excited final state, i.e. the system does not return to its ground state. The final state excitation can be a local (e.g. dd-excitation), including the ligand (charge transfer excitation) or be related to the long-range order of the system (plasmons, magnons, orbitons). The technique is often referred to as (indirect) resonant inelastic X-ray scattering (RIXS).
The general usage in the literature does not strictly distinguish between the two cases (including the present author). RIXS is a second-order process that is theoretically described by the Kramers-Heisenberg equation:
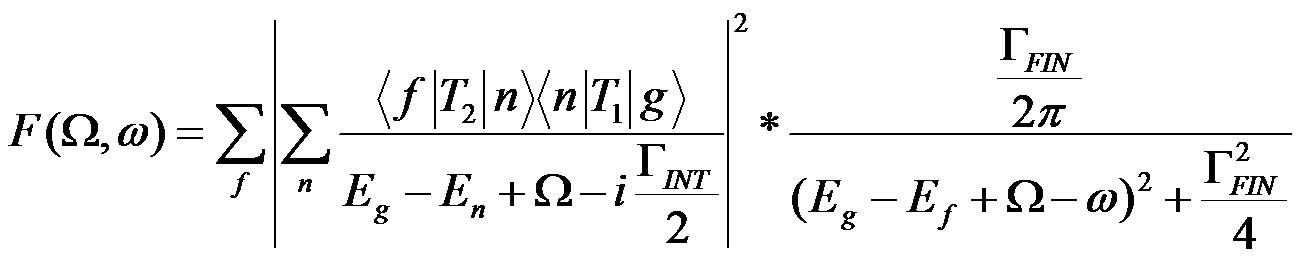
with ground (g), intermediate (n) and final (f) state electron wavefunctions, their energies Eg, En and Ef, the intermediate and final state lifetime broadenings ΓINT and ΓFIN (FWHM) as well as the transition operators T1 and T2 for absorption and emission of an X-ray photon, respectively. The difference between incident and emitted X-ray energy is the energy transfer Ω-ω.
The Kramers-Heisenberg equation is often simplified by neglecting interference effects, multielectron excitations and the angular dependence. We note that non-resonant XES and RIXS can be treated within the same theoretical framework. RIXS allows spectra to be recorded that are only broadened by ΓFIN and not by ΓINT (these are scans recorded at constant incident energy; for details see [1] and [8]). In most cases it holds that ΓINT>ΓFIN and RIXS thus enables the observation of spectral features with sharper line width than in an absorption spectrum. This requires that the instrumental energy band width is sufficiently small. A HERFD scan (vide supra) is NOT only broadened by the final state lifetime. The correct treatment of lifetime and instrumental broadenings have been described by de Groot et al. [23] and Swarbrick et al. [24].
There are major advantages in the application of RIXS/RXES:
- The absorbing atom is not ionized in the case of resonant excitations, as the photoexcited electron stays within a bound state.
- The spectral features become sharper because the lifetime of the intermediate state no longer limits the spectral resolution (this is, in fact, the reason for the sharpening effect in HERFD XAS).
- The final state may not contain a core hole if the energy transfer Ω-ω is sufficiently small. This may greatly facilitate the theoretical analysis and can be used to study core hole effects.
- The final state electronic configuration may formally be equal to other spectroscopies, e.g. the L-edge in 1s2p RIXS of 3d transition metals [20] or UV-Vis in RIXS that exhibits a hole in the valence band in the final state.
When reducing the energy transfer to a few eV one finds new spectral features, some of which have been identified as ligand to metal charge transfer and local dd excitations [21-22]. The energy transfer scale in resonant inelastic X-ray scattering (RIXS) at the valence band is equivalent to optical spectroscopy, but is not limited to a maximum of 6.2 eV as in standard UV-Vis spectroscopy. The selection rules in RIXS are different from UV-Vis because of the two-photon process. As a consequence, the technique is element-selective and optically forbidden transitions may become accessible while the energy resolution is considerably higher in standard UV-Vis spectroscopy.
RIXS spectra require a careful theoretical analysis because of polarization and momentum transfer dependencies. The potential of this technique in the chemical sciences is considerable because valence shell excitations can be observed by means of an element selective, hard X-ray technique.
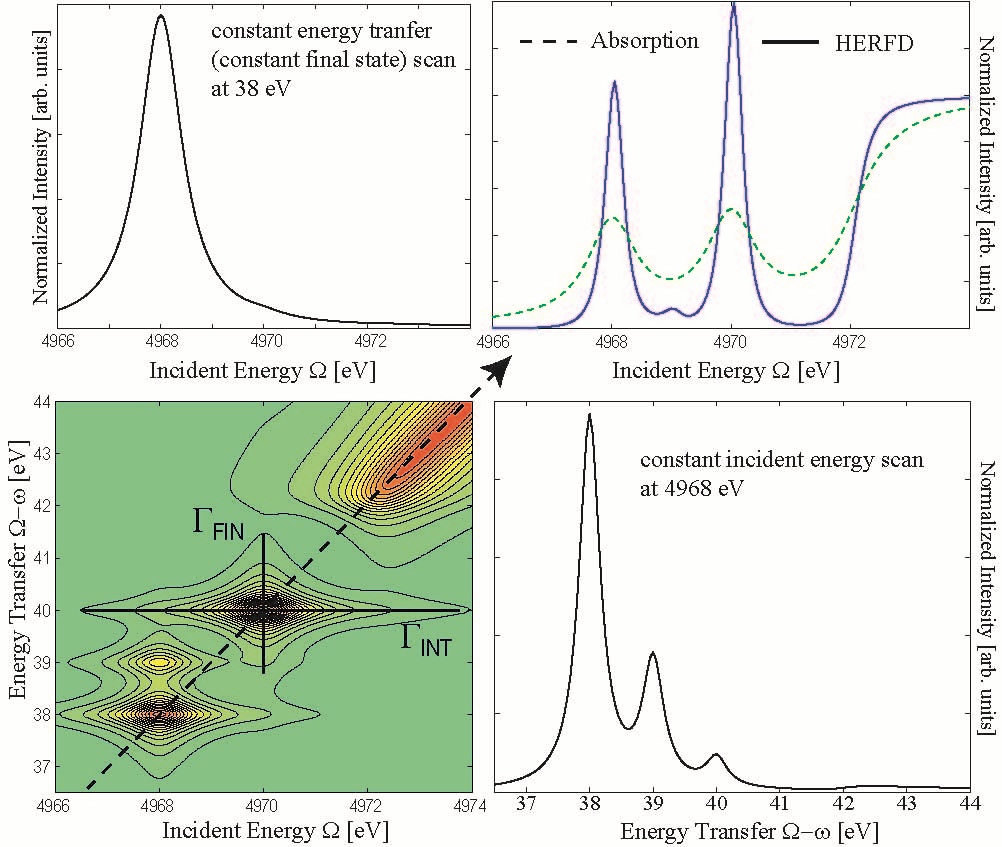
Can we distinguish between localized and delocalized excitations using the Raman shift? This problem is addressed in references [1,8,17]. My current answer to this question is: No. "Localized" refers to the spatial distribution of a one-electron wavefunction. Spectroscopy by definition addresses energy levels. We have the tendency to identify excitations into well isolated (i.e. in energy) orbitals with excitations into spatially localized orbitals. This actually may be the case in some favorable systems but there is to my knowledge no strict theoretical argument to support this relation. The Raman shift (horizontal line in Figure 4 bottom left) occurs for isolated excitations and can easily be understood by analyzing the lifetime broadenings [1]. Coherence is not a prerequisite to observe the Raman shift.
back to topFinal remarks
Instrumentation for photon-out analysis at synchrotron radiation sources has made enormous progress. While research on magnetic materials has been pushing for high energy resolution (ΔE < 100 meV) it is more important for applications in chemistry to capture a large solid angle at moderate energy resolutions of a few hundreds of meV up to a few eV. Research in catalysis, environmental sciences and biology, for example, with low metal concentrations and their need for special sample environments are some of the major research fields to benefit from these developments. Photon-out spectroscopy on systems with less than 0.1 wt% metal concentration is nowadays feasible, and the limit is decreasing with improving instrumentation. The existing in-situ cells for fluorescence detected XAS can be readily installed on an X-ray spectrometer.
References
[2] F.M.F. de Groot and A. Kotani, Core Level Spectroscopy of Solids. Advances in Condensed Matter Science, ed. D.D. Sarma, G. Kotliar, and Y. Tokura. Vol. 6. 2008, New York: Taylor and Francis.
[4] J. Yano, Y. Pushkar, P. Glatzel, A. Lewis, K. Sauer, J. Messinger, U. Bergmann and V. Yachandra, "High-resolution Mn EXAFS of the oxygen-evolving complex in photosystem II: Structural implications for the Mn4Ca cluster", J. Am. Chem. Soc. 127 14974-14975 (2005).
[7] F. Gel’mukhanov and H. Ågren, Physics Reports 312 (1999) 87-330
[12] A. Meisel, G. Leonhardt and R. Szargan, X-Ray Spectra and Chemical Binding. Springer Series in Chemical Physics. Vol. 37. 1989, New York: Springer-Verlag. 458.
[13] J. Messinger, J.H. Robblee, U. Bergmann, C. Fernandez, P. Glatzel, H. Visser, R.M. Cinco, K.L. McFarlane, E. Bellacchio, S.A. Pizarro, S.P. Cramer, K. Sauer, M.P. Klein and V.K. Yachandra, "Absence of Mn-centered oxidation in the S-2 -> S-3 Transition: Implications for the mechanism of photosynthetic water oxidation", J. Am. Chem. Soc. 123 7804-7820 (2001).
[14] J. Badro, G. Fiquet, F. Guyot, J.P. Rueff, V.V. Struzhkin, G. Vanko and G. Monaco, "Iron partitioning in Earth’s mantle: Toward a deep lower mantle discontinuity", Science 300 789-791 (2003).
[16] G.D. Pirngruber, J.D. Grunwaldt, J.A. van Bokhoven, A. Kalytta, A. Reller, O.V. Safonova and P. Glatzel, "On the presence of Fe(IV) in Fe-ZSM-5 and FeSrO3-x - Unequivocal detection of the 3d(4) spin system by resonant inelastic X-ray scattering", J. Phys. Chem. B 110 18104-18107 (2006).
[18] G. Smolentsev, A.V. Soldatov, J. Messinger, K. Merz, T. Weyhermuller, U. Bergmann, Y. Pushkar, J. Yano, V.K. Yachandra, and P. Glatzel, "X-ray Emission Spectroscopy To Study Ligand Valence Orbitals in Mn Coordination Complexes" J. Am. Chem. Soc. (2009) 131 13161-13167; U. Bergmann, C.R. Horne, T.J. Collins, J.M. Workman and S.P. Cramer, "Chemical Dependence of Interatomic X-Ray Transition Energies and Intensities - A Study of Mn K’’ and Kβ2,5 Spectra", Chem. Phys. Lett. 302 119-124 (1999).
[20] F.M.F. de Groot, P. Glatzel, U. Bergmann, P.A. van Aken, R.A. Barrea, S. Klemme, M. Havecker, A. Knop-Gericke, W.M. Heijboer and B.M. Weckhuysen, "1s2p resonant inelastic X-ray scattering of iron oxides", J. Phys. Chem. B 109 20751-20762 (2005).
[24] J.C. Swarbrick, U. Skyllberg, T. Karlsson and P. Glatzel, "High energy resolution X-ray absorption spectroscopy of environmentally relevant lead(II) compounds", Inorg. Chem. 48 10748-10756 (2009).